Introduction
Ensuring plasmid vectors and genes of interest have not mutated in bacterial cell banks can be challenging in molecular cloning workflows. The construction step, where the foreign DNA is inserted into the plasmid vector, may result in the self-re-ligation of the plasmid or incorrect fragment insertion. Although selection gives insight into the presence of a plasmid backbone in a colony after transformation, it does not validate the plasmid content. Large plasmids that require significant cellular resources will increase selective pressures on cells carrying them, causing strains with mutated or partial plasmids to dominate culture. Additional screening is always necessary to ensure the plasmid genetic makeup in your colonies is expected and consistent over time.
Among screening methods, Sanger sequencing stands out for its high accuracy and consistency in identifying the order of nucleotides in DNA sequences. Although next-generation sequencing (NGS) methods are widely used and carry high throughputs, Sanger sequencing remains prominent because of its high accuracy and low cost. and is a preferred method when sequencing the Spike protein in SARS-CoV-2 in applications where NGS is impractical. This article covers principles of Sanger sequencing and how Sanger sequencing can be used to verify the sequence of inserted genes in plasmid DNA constructs.
Sanger methods and the role of ddNTPs
Before understanding the how Sanger sequencing works, it is essential to recall the structure of nucleotides that make up the DNA. A nucleoside triphosphate (dNTP) is composed of a nitrogenous base (A, T, G, or C) attached to a five-carbon sugar residue with a phosphate group. In DNA, a dNTP carries a 3'-OH group that allows it to form a phosphodiester bond with the phosphate group of the next dNTP. DNA polymerase catalyzes bonding between two nucleotides and contributes to the elongation of the DNA sequence.
The termination of a growing DNA strand requires a dideoxynucleotide (ddNTP), which lacks the 3'OH group that prevents the formation of the next phosphodiester bond, preventing further polymerization of the growing DNA strand The function of ddNTPs was first discovered in 1977 by Frederick Sanger and colleagues as a chain termination method for DNA sequencing. More specifically, they used base-specific ddNTPs to determine the base that occupies each position following the primer, revealing the sequence of the bases.
Components of Sanger Sequencing
The following components are necessary for Sanger Sequencing:
- The DNA template to be sequenced, which is obtained from the plasmid through real-time Polymerase Chain Reaction (rtPCR)-based amplification
- A primer complementary to the upstream of the plasmid multiple cloning site (MCS), which is necessary to start the DNA synthesis
- DNA polymerase to start the synthesis
- The four nucleotides or dNTPs, dATP, dTTP, dCTP, and dGTP, which are added to the primer based on the plasmid sequence
- Small amounts of ddNTPs, ddATP, ddTTP, ddCTP, and ddGTP.
Steps
Preparing the Reaction Mixtures
Four reaction mixtures are prepared for classical Sanger sequencing workflows. Each tube contains the plasmid DNA template, the complementary primer, DNA polymerase, the four dNTPs (i.e., dATP, dCTP, dGTP, or dTTP), and a small amount of one of the ddNTPs in each tube.First, the double-stranded plasmid DNA is denatured to form a single-stranded template.
Chain Termination
In each tube, DNA polymerase synthesizes and extends DNA strands by sequentially adding one of the four dNTPs. However, when a ddNTP gets incorporated, the DNA synthesis is terminated since no further dNTPs can be linked. For instance, in the tube containing ddATP, chain termination may occur at every location where dATP would normally be integrated. Because there are a small amount of ddATP nucleotides relative to dATP nucleotides, polymerization reactions will continue until a ddATP nucleotide is selected at random, leading to polymerization reactions being terminated at varying lengths. Repeating this reaction many times ensures that ddATP-termination occurs at every possible position. Eventually, DNA fragments of varying lengths, each terminated with a ddATP, are generated in this tube. By the same logic, each tube will contain mixtures of varying DNA fragments terminated by the specific ddNTP spiked in.
Separation: Radioactive Labeling vs Fluorescent Labeling
Next, the DNA fragments can be separated according to their lengths.
In initial Sanger sequencing procedures, this was achieved by radioactive labeling of the ddNTPs and running the DNA fragments through polyacrylamide gel electrophoresis. Each respective ddNTP reaction is run in its own lane. The fragments from each tube were separately loaded onto one of the four lanes. The shorter a fragment, the farther it will travel in the polyacrylamide gel. Autoradiography was performed to visualize the radioactively labeled fragments and deduce the sequence of the complementary DNA template. For example, the lane containing ddATP-terminated fragments reveals each position relative to the primer where A was added to complement a T in the DNA template. Reading the gel inclusive of all four lanes from bottom to top reveals the entire sequence of the DNA template in order, which could be used to confirm correct gene insertion in the plasmid DNA. While radioactive labeling used to be essential for Sanger sequencing, this method has fallen out of favor.
Fluorescent Detection and Analysis
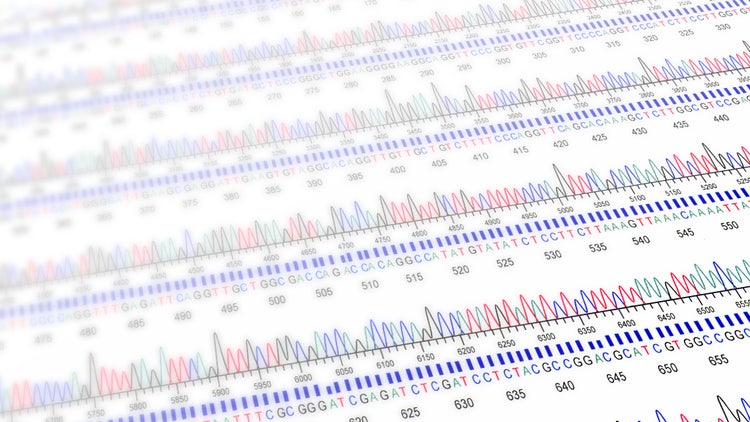
Modern Sanger sequencing entails fluorescent labeling of the ddNTPs instead of radiolabeling. Each ddNTP is tagged with a fluorophore emitting a specific color. The unique fluorescent labeling of ddNTPs allows all reactions to be performed in the same tube. The resulting DNA fragments can be run through a single lane, enabling the employment of a more advanced separation workflow via capillary electrophoresis.
Fluorescent labeling of ddNTPs drives automated sequence detection workflows, making it more advantageous than traditional radiolabeling methods. Here, a laser detector is integrated into the capillary gel system, illuminating each passing fragment by exciting the fluorophore on its terminal ddNTP. The shorter fragments reach the detector faster and are recorded first. The order of fluorescent colors is recorded and computationally parsed into a chromatogram, which is used to determine the target DNA sequence and compare it to a reference sequence to confirm successful gene insertion.
See how Danaher Life Sciences can help
Advantages
Sanger sequencing maintains its gold standard state for molecular cloning and research applications, owing to several advantages.
- It is highly accurate in sequence validation.
- Fluorescent labeling and capillary gel electrophoresis yield consistent and reliable sequencing results.
- Sanger sequencing is easy to perform, as it does not rely on complex bioinformatics software for output interpretation. The chromatograms obtained from separation are high-quality and easy to interpret.
- Sanger sequencing is particularly useful and cost-effective for small-scale projects involving the sequencing of short insertions.
- It can read up to 1000 base pairs, which is longer than what next-generation sequencing (NGS) provides without downstream computing workflows.
Challenges
One of the main challenges in Sanger sequencing is the low throughput, as it can only sequence fragments between 300-1000 base pairs. This method is more suitable for specifically sequencing the insert regions and may not effectively help identify structural issues in the whole plasmid in a single run.
Sanger sequencing does not allow simultaneous sequencing of multiple genes in a single reaction, making the sequencing of larger DNA fragments and multiple samples time-consuming. Specific primers must be designed for each sequencing run. It also requires high-quality and pure DNA samples, so contamination during plasmid DNA isolation can profoundly affect quality of final results.
Additionally, the first 15-40 bases of sequencing are not always accurate due to non-specific primer binding. It is essential to use a custom primer that is at least 50 base pairs upstream of the gene of interest.
Featured Product
Sanger Sequencing for Plasmid DNA Verification
GenomeLab GeXP™ System
Chosen because it is a capillary electrophoresis-based genetic analysis system used for multiplexed gene expression profiling, fragment analysis and sequencing QC, tying directly to this page's subject of sanger sequencing plasmid dna verification.

DNA Plasmid Purification
Chosen as a complementary second option because it is the CosMCPrep bead-based reagent kit for purifying high- and low-copy plasmid, cosmid and BAC DNA, also directly relevant to sanger sequencing plasmid dna verification.

See how Danaher Life Sciences can help
FAQs
How does Sanger sequencing compare to next-generation sequencing (NGS) for plasmid screening?
1. Accuracy: Sanger sequencing provides high accuracy for small regions but may struggle with repetitive or GC-rich sequences. NGS offers high accuracy over the entire plasmid.
2. Throughput: Sanger sequencing is low-throughput and suitable for a few plasmids, while NGS can screen hundreds to thousands of plasmids simultaneously.
3. Cost: Sanger sequencing is more cost-effective for small-scale projects, whereas NGS is economical for large-scale screening.
4. Data Complexity: Sanger sequencing produces straightforward, single-read data, while NGS generates large datasets requiring advanced bioinformatics tools for analysis.
What are the main applications of Sanger sequencing in plasmid screening?
1. Verification of Cloning: Confirming the presence and sequence of inserted DNA.
2. Mutation Detection: Screening for point mutations or engineered changes.
3. Quality Control: Ensuring plasmid integrity before downstream applications like protein expression or gene delivery.
How can you improve the success rate of Sanger sequencing for plasmid DNA?
1. Use high-quality, purified plasmid DNA.
2. Design primers with optimal melting temperature (Tm) and minimal secondary structures.
3. Avoid sequencing GC-rich regions in a single reaction by using overlapping primers.
4. Optimize DNA concentration to avoid signal artifacts.
5. Use a sequencing service with experience handling plasmid DNA.
What limitations should be considered when using Sanger sequencing for plasmid screening?
1. Limited read length (~700–900 bases), which may require multiple primers and reactions for large plasmids.
2. Difficulty in sequencing regions with homopolymers, strong secondary structures, or high GC content.
3. Lower throughput compared to high-throughput methods like NGS.